
Nathan Teuscher and Nikunjkumar Patel , Certara03.07.17
Modeling and simulation (M&S) has already profoundly impacted drug development and formulation. It is used in 90% of all U.S. Food and Drug Administration (FDA) drug approvals. M&S can influence every phase of the drug development process, including informing commercial decisions about the benefits of bringing a specific drug to market. It can be employed to compare the safety and efficacy of drug candidates; select dose and dose regimen; and identify potential drug-drug and drug-food interactions. M&S can be used in lieu of specific clinical trials, and of importance to those involved in drug formulation, leveraged to demonstrate virtual bioequivalence and obtain biowaivers.
Formulation science is an iterative process that can benefit from the ‘predict, learn, confirm and apply’ paradigm underpinning M&S. That paradigm can support and strengthen the overall formulation strategy and inform the numerous alternate formulations that will be developed throughout the development cycle. Formulation selection can profoundly impact drug release, absorption, and metabolism, altering the drug’s pharmacokinetic profile and its pharmacodynamic response.
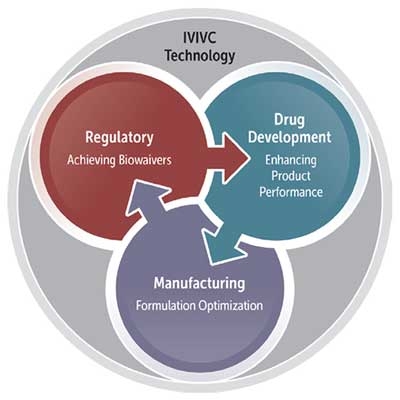
In short, M&S can predict the in vivo performance of drug products. Therefore, its use can improve formulation strategy by aiding scientists in designing a rational and cost-effective approach to formulation development (see Figure 1).
What is modeling and simulation?
M&S is an integrative science, which incorporates relationships between disease and biological pathways, drug characteristics, and individual variability, and leverages existing knowledge to guide future research. M&S is an invaluable drug development and formulation tool whose use is actively encouraged by global regulators.
An in-vitro in-vivo correlation (IVIVC) has been defined by the FDA as “a predictive mathematical model describing the relationship between an in-vitro property of a dosage form and an in-vivo response.” The U.S. Pharmacopoeia (USP) defines IVIVC as establishing a rational relationship between a biological property produced by a dosage form, and a physiochemical property of the same dosage form.
Developing and optimizing a new drug formulation may involve changes in the drug composition, manufacturing process, use and type of equipment, or batch size. These changes, which can occur often, trigger the need to conduct bioavailability studies to demonstrate ‘equivalence’ between the original accepted formulation and the new one. IVIVC is a biopharmaceutical tool used in drug development and formulation optimization to demonstrate that equivalence.
IVIVC: Accelerating formulation development and saving costly studies
IVIVC technology allows formulation and manufacturing professionals to understand how dissolution parameters affect in vivo drug performance. Dissolution testing is required for all solid oral dosage forms and is used in all phases of development for product release and stability testing. This key analytical test can detect physical changes in an active pharmaceutical ingredient and in the formulated product.
This information can be determined quickly, under inexpensive, controlled lab conditions, to serve as a surrogate for the in vivo drug behavior rather than through expensive and time-consuming animal or human bioavailability or bioequivalence (BA or BE) studies. Each BA/BE study replaced by IVIVC analysis can shorten the development cycle by months and save hundreds of thousands of dollars. At the same time, IVIVC gives scientists the requisite knowledge to tweak a drug’s formulation to improve its in vivo performance with fewer iterations and false starts.
There are many instances during a drug’s life cycle when its bioequivalence may need to be established. Its formulation may have been changed to alter its release timing, improve its solubility and absorption, reduce manufacturing costs, extend its shelf life, prevent adverse events such as an upset stomach, improve its taste or smell, or hold the tablet together better. New formulations may be required multiple times during a drug’s life cycle because of changes to the manufacturing supplies, processes, dosage forms, or other factors. In many of these instances, IVIVC can be used to gain a biowaiver for the product.
Modeling and simulation for IVIVC
There are two main types of M&S for IVIVC, which when used in a complementary way are considered ‘best practice’.
Conventional (statistical) IVIVC. This method uses deconvolution methods such as Wagner-Nelson, Loo-Riegelman, numerical deconvolution and modified deconvolution to estimate the rate of input of a drug into the systemic circulation of observed plasma drug concentrations from the oral formulation.
The FDA guidance for, “Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations,” mentions four levels of IVIVC (A, B, C, and D) based on the correlation’s ability to reflect accurately the complete plasma drug concentration-time profile resulting from administering the given dosage form. Level A represents the highest correlation—a point-to-point statistical relationship between the in vitro dissolution rate and in vivo input rate. This approach is used most often to attain biowaivers.
While conventional IVIVC is the gold standard for drugs displaying a linear relationship between absorption and dissolution, today’s more complex drugs need alternative modeling approaches. FDA held a workshop in May 2016 where it brought together experts from industry, academia and the regulatory agency to address this need and evaluate mechanistic IVIVC as an evolving technology for drug formulation. During that workshop, the FDA shared its current thinking on mechanism-based absorption M&S and obtained public input on its future application for bioequivalence determination and other regulatory activities for oral drug products.
Mechanistic IVIVC (PBPK). This approach considers separately the various mechanisms involved in drug absorption, such as transit time, gut wall permeability, gut wall metabolism, and hepatic first-pass metabolism from dissolution rate. By integrating the anatomical and physiological parameters of the gastrointestinal (GI) tract with the physicochemical properties of drug substances, mechanistic IVIVC provides scientists with valuable insights for designing and evaluating the performance and safety of new drug formulations. Mechanistic models provide them with a detailed understanding of the mechanisms involved in absorption and how critical they are for formulation.
Mechanistic physiologically-based pharmacokinetic (PBPK) models, such as the Simcyp Simulator, can be used to estimate in vivo dissolution rather than just the systemic input rate, separately accounting for permeation, GI transit and first-pass elimination. This enables the IVIVC to be integrated with other model components to predict in vivo product performance across a specific patient population. Information on transporters and drug metabolic pathways can also be incorporated to model in vivo exposure for a specific body part or the blood.
How to select the most appropriate IVIVC modeling method
Oral drugs can be classified according to the Biopharmaceutics Classification System (BCS), which is based on the drug’s solubility and permeability. Established in the 1990s and adopted by the FDA in 2000, the BCS provides a regulatory framework for new drug formulations that includes setting dissolution specifications, supporting risk assessment, evaluating post-approval changes, and approving biowaivers. Additionally, the BCS provides a framework for drug development starting with candidate selection and moving to pre-formulation evaluation, solid form selection, and formulation strategies.
The body’s ability to absorb oral drug products varies according to physiological and physiochemical factors and the dosage form. The main physiological processes impacting in vivo dissolution and absorption within the GI tract are secretion, digestion, and absorption. Underlying those processes are factors including pH; fluid, electrolyte, peptide, protein and digestive enzyme levels; surface area; GI motility and transit time; and bile secretion. The key physiochemical properties of drug molecules are solid particle dissolution, ionization, and solubility. When evaluating dosage form, whether it be immediate, modified, extended or delayed release, the rate and amount of absorption are influenced by the drug design and dissolution profile.
The IVIVC method selected depends on the type of product and the objectives of the modeling. With numerical approaches, inter- and intra-subject variability is noise. Including or excluding statistical covariates and/or modifying the study design can minimize that noise. The goal is correlating the in vivo and in vitro data.
In contrast, when using a mechanistic approach, separating drug and formulation parameters from subject physiology variability allows associated variabilities to be estimated and projected. This provides an opportunity for population-level simulation and analysis. It also allows scientists to extrapolate from one physiological condition to another, allowing them to answer ‘what if’ questions or virtually assess untested clinical scenarios. Assuming that the IVIVC remains valid for alternate formulations or with other patient physiologies, scientists can expand their product understanding beyond the limits of the available IVIVC dataset and migrate into personalized medicine.
Many new drug candidates are poorly soluble, which can severely limit their bioavailability. To ameliorate this issue, a widely used approach is formulating supersaturated drug solutions. However, supersaturated solutions risk precipitation, which can limit the intended benefits of this approach. To address this issue, the U.S. FDA recently awarded Certara’s Simcyp division a grant to develop a mechanistic modeling and simulation framework to predict the behavior of orally-dosed supersaturating drugs and drug products (see Figure 2).
Strategic value of M&S in formulation
The U.S. FDA, European Medicines Agency (EMA), Japanese Pharmaceuticals and Medical Devices Agency, and other global regulatory agencies encourage the use of IVIVC. They consider it an important drug development tool, which enhances product and process understanding, with the ultimate goal of ensuring consistent performance throughout the product’s life cycle.
FDA recently published a paper, entitled, “Regulatory Experience with In Vivo In Vitro Correlations in New Drug Applications.” The authors discussed using IVIVC for pre-approval (bridging formulations to the pivotal BA/BE batch), and post-approval (formulation, manufacturing site, and process) changes to reduce the need for in vivo bioequivalence studies.
They also stressed the value that IVIVC can provide by facilitating decision making during drug development, especially in light of the Quality by Design (QbD) paradigm. QbD, promoted by FDA and EMA, is the concept that product and process performance characteristics are scientifically designed to meet specific objectives, not merely empirically derived from performance of test batches. IVIVC is leveraged for three purposes:
The challenge was developing an IVIVC model for a compound and formulation whose release rate, specific bioavailability, and non-linear relationships would not initially seem to support an IVIVC.
The formulation under evaluation was a capsule containing four different types of delayed release beads. Each bead was characterized by a mean dissolution time (MDT) and a time lag (Tlag) and each of the seven formulations under study was comprised of a different ratio of each bead type.
The data simulation model contained elements of stomach emptying, intestinal and colon transit, first-pass metabolism in the small intestine and multi-compartment pharmacokinetics. The simulations were done in three stages: simulating dissolution profiles, generating subject PK parameters from a multivariate log-normal distribution, and simulating individual plasma profiles using three kinetic compartments, two absorption compartments with transit times, and proportional error on observations.
Using conventional IVIVC methodology with Phoenix IVIVC software, mean plasma profiles for four formulations—10-20 subjects per formulation—were deconvolved using the PK parameters relating to the intravenous kinetics of the compound. The generated series of fraction-absorbed profiles were then overlaid with the dissolution profiles. Reviewing this plot suggested that the slower formulations had greater bioavailability than the fastest release, and the slower the release, the greater the bioavailability.
This meant that a simple model could not describe the data well, requiring a custom model to fit the nonlinearity of the compound. Since the Levy plot indicated two time scales, a bi-linear model was used with an initial lag consistent with a stomach emptying effect together with data representing intestinal transit time switching between phases.
Inferences drawn from data exploration can guide IVIVC development. In this case, an initial model was proposed after analyzing two exploratory plots. Model refinement is motivated by consideration of physiological mechanisms and leads to a suitable IVIVC, used for regulatory purposes.
Case Study: IVIVC using physiologically-based absorption modeling
This project used mechanistic modeling to establish IVIVC for a controlled release formulation of topiramate. Topiramate’s bioavailability depends on the site of GI absorption. Specifically, topiramate exhibits higher bioavailability in the colon compared to the stomach. For example, a dosage can be designed wherein the delayed-release component contains less topiramate than the immediate-release component but that nonetheless achieves a blood plasma concentration equivalent to the immediate-release component. Alternatively, a dosage can be designed so that the delayed-release component contains an amount of topiramate equal to that of the immediate-release component yet would achieve a blood plasma concentration greater than that of the immediate-release component.
To study this, a PBPK model was built using the Simcyp Advanced Dissolution Absorption and Metabolism (ADAM) model. Four formulations—slow, medium and fast release and oral—and multiple immediate release doses were studied, using clinical dataset, formulation and food-drug interaction data. The PBPK approach improved the predictive performance of the IVIVC model, producing a linear IVIVC.
The discovery of the differential bioavailability of topiramate allows a more rational drug design based upon the specific therapeutic profiles to be achieved. IVIVC modeling facilitates these alternative formulations.
Conclusion
IVIVC M&S plays a strategic role throughout the drug development and formulation process. It allows sponsors to replace expensive, time-consuming BA/BE studies with more efficient, inexpensive IVIVC analyses, shaving months and hundreds of thousands of dollars off development. It also provides scientists with greater product insight, allowing them to change a drug’s formulation to improve both its in vivo performance and likelihood of regulatory success.
Nathan Teuscher, Ph.D., is the vice president of pharmacometric solutions at Certara.
Nikunjkumar Patel, M.S. (Pharm), is a senior research scientist in Certara’s modelling and simulations group.
Formulation science is an iterative process that can benefit from the ‘predict, learn, confirm and apply’ paradigm underpinning M&S. That paradigm can support and strengthen the overall formulation strategy and inform the numerous alternate formulations that will be developed throughout the development cycle. Formulation selection can profoundly impact drug release, absorption, and metabolism, altering the drug’s pharmacokinetic profile and its pharmacodynamic response.
In short, M&S can predict the in vivo performance of drug products. Therefore, its use can improve formulation strategy by aiding scientists in designing a rational and cost-effective approach to formulation development (see Figure 1).
What is modeling and simulation?
M&S is an integrative science, which incorporates relationships between disease and biological pathways, drug characteristics, and individual variability, and leverages existing knowledge to guide future research. M&S is an invaluable drug development and formulation tool whose use is actively encouraged by global regulators.
An in-vitro in-vivo correlation (IVIVC) has been defined by the FDA as “a predictive mathematical model describing the relationship between an in-vitro property of a dosage form and an in-vivo response.” The U.S. Pharmacopoeia (USP) defines IVIVC as establishing a rational relationship between a biological property produced by a dosage form, and a physiochemical property of the same dosage form.
Developing and optimizing a new drug formulation may involve changes in the drug composition, manufacturing process, use and type of equipment, or batch size. These changes, which can occur often, trigger the need to conduct bioavailability studies to demonstrate ‘equivalence’ between the original accepted formulation and the new one. IVIVC is a biopharmaceutical tool used in drug development and formulation optimization to demonstrate that equivalence.
IVIVC: Accelerating formulation development and saving costly studies
IVIVC technology allows formulation and manufacturing professionals to understand how dissolution parameters affect in vivo drug performance. Dissolution testing is required for all solid oral dosage forms and is used in all phases of development for product release and stability testing. This key analytical test can detect physical changes in an active pharmaceutical ingredient and in the formulated product.
This information can be determined quickly, under inexpensive, controlled lab conditions, to serve as a surrogate for the in vivo drug behavior rather than through expensive and time-consuming animal or human bioavailability or bioequivalence (BA or BE) studies. Each BA/BE study replaced by IVIVC analysis can shorten the development cycle by months and save hundreds of thousands of dollars. At the same time, IVIVC gives scientists the requisite knowledge to tweak a drug’s formulation to improve its in vivo performance with fewer iterations and false starts.
There are many instances during a drug’s life cycle when its bioequivalence may need to be established. Its formulation may have been changed to alter its release timing, improve its solubility and absorption, reduce manufacturing costs, extend its shelf life, prevent adverse events such as an upset stomach, improve its taste or smell, or hold the tablet together better. New formulations may be required multiple times during a drug’s life cycle because of changes to the manufacturing supplies, processes, dosage forms, or other factors. In many of these instances, IVIVC can be used to gain a biowaiver for the product.
Modeling and simulation for IVIVC
There are two main types of M&S for IVIVC, which when used in a complementary way are considered ‘best practice’.
Conventional (statistical) IVIVC. This method uses deconvolution methods such as Wagner-Nelson, Loo-Riegelman, numerical deconvolution and modified deconvolution to estimate the rate of input of a drug into the systemic circulation of observed plasma drug concentrations from the oral formulation.
The FDA guidance for, “Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations,” mentions four levels of IVIVC (A, B, C, and D) based on the correlation’s ability to reflect accurately the complete plasma drug concentration-time profile resulting from administering the given dosage form. Level A represents the highest correlation—a point-to-point statistical relationship between the in vitro dissolution rate and in vivo input rate. This approach is used most often to attain biowaivers.
While conventional IVIVC is the gold standard for drugs displaying a linear relationship between absorption and dissolution, today’s more complex drugs need alternative modeling approaches. FDA held a workshop in May 2016 where it brought together experts from industry, academia and the regulatory agency to address this need and evaluate mechanistic IVIVC as an evolving technology for drug formulation. During that workshop, the FDA shared its current thinking on mechanism-based absorption M&S and obtained public input on its future application for bioequivalence determination and other regulatory activities for oral drug products.
Mechanistic IVIVC (PBPK). This approach considers separately the various mechanisms involved in drug absorption, such as transit time, gut wall permeability, gut wall metabolism, and hepatic first-pass metabolism from dissolution rate. By integrating the anatomical and physiological parameters of the gastrointestinal (GI) tract with the physicochemical properties of drug substances, mechanistic IVIVC provides scientists with valuable insights for designing and evaluating the performance and safety of new drug formulations. Mechanistic models provide them with a detailed understanding of the mechanisms involved in absorption and how critical they are for formulation.
Mechanistic physiologically-based pharmacokinetic (PBPK) models, such as the Simcyp Simulator, can be used to estimate in vivo dissolution rather than just the systemic input rate, separately accounting for permeation, GI transit and first-pass elimination. This enables the IVIVC to be integrated with other model components to predict in vivo product performance across a specific patient population. Information on transporters and drug metabolic pathways can also be incorporated to model in vivo exposure for a specific body part or the blood.
How to select the most appropriate IVIVC modeling method
Oral drugs can be classified according to the Biopharmaceutics Classification System (BCS), which is based on the drug’s solubility and permeability. Established in the 1990s and adopted by the FDA in 2000, the BCS provides a regulatory framework for new drug formulations that includes setting dissolution specifications, supporting risk assessment, evaluating post-approval changes, and approving biowaivers. Additionally, the BCS provides a framework for drug development starting with candidate selection and moving to pre-formulation evaluation, solid form selection, and formulation strategies.
The body’s ability to absorb oral drug products varies according to physiological and physiochemical factors and the dosage form. The main physiological processes impacting in vivo dissolution and absorption within the GI tract are secretion, digestion, and absorption. Underlying those processes are factors including pH; fluid, electrolyte, peptide, protein and digestive enzyme levels; surface area; GI motility and transit time; and bile secretion. The key physiochemical properties of drug molecules are solid particle dissolution, ionization, and solubility. When evaluating dosage form, whether it be immediate, modified, extended or delayed release, the rate and amount of absorption are influenced by the drug design and dissolution profile.
The IVIVC method selected depends on the type of product and the objectives of the modeling. With numerical approaches, inter- and intra-subject variability is noise. Including or excluding statistical covariates and/or modifying the study design can minimize that noise. The goal is correlating the in vivo and in vitro data.
In contrast, when using a mechanistic approach, separating drug and formulation parameters from subject physiology variability allows associated variabilities to be estimated and projected. This provides an opportunity for population-level simulation and analysis. It also allows scientists to extrapolate from one physiological condition to another, allowing them to answer ‘what if’ questions or virtually assess untested clinical scenarios. Assuming that the IVIVC remains valid for alternate formulations or with other patient physiologies, scientists can expand their product understanding beyond the limits of the available IVIVC dataset and migrate into personalized medicine.
Many new drug candidates are poorly soluble, which can severely limit their bioavailability. To ameliorate this issue, a widely used approach is formulating supersaturated drug solutions. However, supersaturated solutions risk precipitation, which can limit the intended benefits of this approach. To address this issue, the U.S. FDA recently awarded Certara’s Simcyp division a grant to develop a mechanistic modeling and simulation framework to predict the behavior of orally-dosed supersaturating drugs and drug products (see Figure 2).
Strategic value of M&S in formulation
The U.S. FDA, European Medicines Agency (EMA), Japanese Pharmaceuticals and Medical Devices Agency, and other global regulatory agencies encourage the use of IVIVC. They consider it an important drug development tool, which enhances product and process understanding, with the ultimate goal of ensuring consistent performance throughout the product’s life cycle.
FDA recently published a paper, entitled, “Regulatory Experience with In Vivo In Vitro Correlations in New Drug Applications.” The authors discussed using IVIVC for pre-approval (bridging formulations to the pivotal BA/BE batch), and post-approval (formulation, manufacturing site, and process) changes to reduce the need for in vivo bioequivalence studies.
They also stressed the value that IVIVC can provide by facilitating decision making during drug development, especially in light of the Quality by Design (QbD) paradigm. QbD, promoted by FDA and EMA, is the concept that product and process performance characteristics are scientifically designed to meet specific objectives, not merely empirically derived from performance of test batches. IVIVC is leveraged for three purposes:
- Regulatory: As a surrogate for in vivo bioequivalence studies and to support attaining biowaivers; in lieu of certain in vivo experiments; to evaluate pre- and post-approval manufacturing changes; and to evaluate custom risk factors to guide post-marketing surveillance.
- Drug development: To provide initial guidance and direction for early formulation development; to inform how and what is chosen for the in vitro release testing methods; to help understand PK variability so that scientists can design better bioequivalent studies and reduce sample sizes; to extrapolate data from healthy volunteers in BE studies to patients with GI conditions; to prioritize formulation efforts pertaining to product development, and oftentimes minimize the number of pilot studies required; to enhance drug product understanding during the development cycle; and to reduce regulatory uncertainty and gain product approval more quickly.
- Formulation and manufacturing: To evaluate potential performance differences for modified release formulations; to test the impact of each component and assure quality control in formulation and reformulation; to reduce the number of bioequivalence studies required due to scale-up and post-marketing changes; and to bridge formulations to the pivotal BA/BE batch.
The challenge was developing an IVIVC model for a compound and formulation whose release rate, specific bioavailability, and non-linear relationships would not initially seem to support an IVIVC.
The formulation under evaluation was a capsule containing four different types of delayed release beads. Each bead was characterized by a mean dissolution time (MDT) and a time lag (Tlag) and each of the seven formulations under study was comprised of a different ratio of each bead type.
The data simulation model contained elements of stomach emptying, intestinal and colon transit, first-pass metabolism in the small intestine and multi-compartment pharmacokinetics. The simulations were done in three stages: simulating dissolution profiles, generating subject PK parameters from a multivariate log-normal distribution, and simulating individual plasma profiles using three kinetic compartments, two absorption compartments with transit times, and proportional error on observations.
Using conventional IVIVC methodology with Phoenix IVIVC software, mean plasma profiles for four formulations—10-20 subjects per formulation—were deconvolved using the PK parameters relating to the intravenous kinetics of the compound. The generated series of fraction-absorbed profiles were then overlaid with the dissolution profiles. Reviewing this plot suggested that the slower formulations had greater bioavailability than the fastest release, and the slower the release, the greater the bioavailability.
This meant that a simple model could not describe the data well, requiring a custom model to fit the nonlinearity of the compound. Since the Levy plot indicated two time scales, a bi-linear model was used with an initial lag consistent with a stomach emptying effect together with data representing intestinal transit time switching between phases.
Inferences drawn from data exploration can guide IVIVC development. In this case, an initial model was proposed after analyzing two exploratory plots. Model refinement is motivated by consideration of physiological mechanisms and leads to a suitable IVIVC, used for regulatory purposes.
Case Study: IVIVC using physiologically-based absorption modeling
This project used mechanistic modeling to establish IVIVC for a controlled release formulation of topiramate. Topiramate’s bioavailability depends on the site of GI absorption. Specifically, topiramate exhibits higher bioavailability in the colon compared to the stomach. For example, a dosage can be designed wherein the delayed-release component contains less topiramate than the immediate-release component but that nonetheless achieves a blood plasma concentration equivalent to the immediate-release component. Alternatively, a dosage can be designed so that the delayed-release component contains an amount of topiramate equal to that of the immediate-release component yet would achieve a blood plasma concentration greater than that of the immediate-release component.
To study this, a PBPK model was built using the Simcyp Advanced Dissolution Absorption and Metabolism (ADAM) model. Four formulations—slow, medium and fast release and oral—and multiple immediate release doses were studied, using clinical dataset, formulation and food-drug interaction data. The PBPK approach improved the predictive performance of the IVIVC model, producing a linear IVIVC.
The discovery of the differential bioavailability of topiramate allows a more rational drug design based upon the specific therapeutic profiles to be achieved. IVIVC modeling facilitates these alternative formulations.
Conclusion
IVIVC M&S plays a strategic role throughout the drug development and formulation process. It allows sponsors to replace expensive, time-consuming BA/BE studies with more efficient, inexpensive IVIVC analyses, shaving months and hundreds of thousands of dollars off development. It also provides scientists with greater product insight, allowing them to change a drug’s formulation to improve both its in vivo performance and likelihood of regulatory success.
Nathan Teuscher, Ph.D., is the vice president of pharmacometric solutions at Certara.
Nikunjkumar Patel, M.S. (Pharm), is a senior research scientist in Certara’s modelling and simulations group.